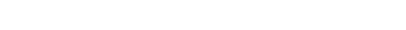
Application and development of methods for the calculation of the electronic structure of polymers
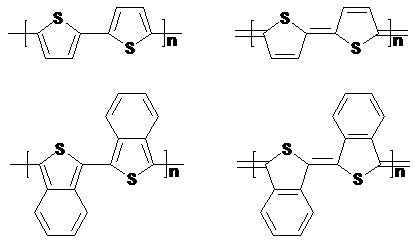
The calculation of the electronic structure of polymers can be achieved through the oligomer approach or K-space methods. Both approaches have various advantages in the employ both depending on the problem at hand. Currently density functional theory is the most effective and accurate approach. In our testing focus on obtaining reliable predictions for the geometry and the electronic structure. We are interested in systems which often display valance tautomerism where electronic structure depends sensitively on the structural parameters as illustrated for the two forms of polydiacetylene above. In such cases the choice of method is essential. We shown that with proper choice of the exact exchange content in hybrid density functional theory this goal can be achieved with high accuracy. Further plans in this direction include the search for proper levels of theory that include van der Waals interactions.
Selected references:
- “Bandgap Calculations for Conjugated Polymers”
Yang, S.; Olishevski, P.; Kertesz, M. Synth Met. 2004, 141, 171–177. - “Conjugated Polymers and Aromaticity”
Kertesz, M.; Choi, C.H.; Yang, S.J. Chem. Rev., 2005, 105(10), 3448 - 3481. - “Energy gaps and their control in thiophene based polymers and oligomers”, (invited review)
Miklos Kertesz, Shujiang Yang, and Yonghui Tian, in “Thiophene-Based Materials for Electronics and Photonics”, Edited by I. Perepichka and D. Perepichka, Wiley, 2009. (pp. 341-365.)